Joint Clinical Assessment (JCA) provides an opportunity for manufacturers and Health Technology Assessment (HTA) bodies to work more closely with one another; but what do these opportunities look like? And will there be an equal number of challenges to overcome? Clarivate’s HTA and RWE experts recently explored this at the Evidence, Pricing and Access Conference in Amsterdam (12-13th March). We covered JCA essentials, including when it is coming and what it might mean for medical innovation in different therapeutic categories (e.g. oncology, rare disease and immunotherapies). We also explored questions around how we might build a roadmap to effective JCA engagement and outcomes –- what will ‘good’ look like? How can we leverage real world evidence (RWE) to support the new JCA process, optimize access and ensure new products reach the right patients as early as possible?
What is JCA and when is it coming?
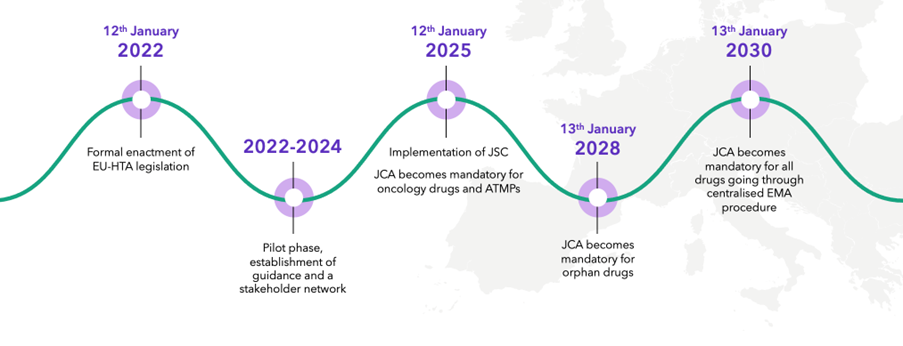
JCA is a centralized European procedure to evaluate the clinical evidence for a product as part of the HTA process. It was formally enacted on January 12, 2022, with the pilot phase and establishment of guidance and stakeholder network being put into place over the last couple of years.
There are, however, still tenders out by the E.U. commission for provision of training of HTABs in how to manage the new process, and there have been a few delays in the overall timing of some elements of the process. JCA becomes mandatory for oncology products and ATMPs on January 12, 2025, for orphan drugs in 2028 and will become mandatory for all drugs going through the European Medicines Agency (EMA) procedure on January 13, 2030.
JCA implementation: key dates
Source: Clarivate
Four key challenges for pharmas in JCA
While the purpose of JCA is to expedite patient access to new treatments by promoting a more efficient and coordinated HTA assessment process, it brings several challenges for manufacturers.
Firstly, the JCA dossier will need to be submitted by companies at around Day 170 of the process, which is before the EMA releases the list of outstanding issues (at Day 180) and before the CHMP opinion (at Day 210). Submitting a dossier without knowing the final population will undoubtedly be challenging for manufacturers as the label (including the indication) will not be final, and may even require the re-start of JCA. In addition, there will only be 90 days between confirmation of the Population, Intervention, Comparator(s), Outcomes (PICOs) and dossier submission, leaving little time to pull together a comprehensive and high-quality evidence dossier without significant preplanning. Furthermore, the submission is likely to happen far in advance of economic discussions and pricing negotiations in individual countries, making it hard to plan strategically for optimal access and reimbursement in each country.
Secondly, the scope of the assessment is a challenge. Even with reassurances in the most recent Implementation Act, with promises of consolidated PICOs as far as possible, it is still likely that for some treatments, there is potential for many PICOs, and this may prove exceedingly difficult for the manufacturer. PICOs will potentially differ where countries consider different relevant comparators because they have different standards of care. They may also define subgroups and surrogate endpoints differently. This is likely to result in significant challenges during the PICO stage of the assessment, especially in determining a clear strategy and narrative and in generating evidence (in the format of meta-analyses, for example) in a timely manner. This is made even more challenging by the apparent lack of manufacturer involvement in this part of the process.
Thirdly, different countries may have different data requirements. Manufacturers will have to address this regardless of JCA. However, we note that JCA does not mean that countries cannot request additional data, and in some situations, it may not be the more efficient process that is promised. Indeed, compiling a dossier for JCA and then responding to requests from several countries (within a 7-to-30-day window) may increase the manufacturer’s workload in some cases.
Finally, we have worked on enough HTA submissions to know that it is not just about putting the clinical and economic data into a dossier and hoping for the best. Successful HTA submissions have a clear narrative running through background, clinical, and economic sections, which is typically the result of months of planning and strategic discussion. While some countries do already have separate clinical and economic dossiers (and even when there is a single dossier, reviewers may only focus on the sections relevant to them), it comes back to the timing -– creating that convincing narrative that runs through all sections of a submission will be made harder by the much earlier submission of the background and clinical data.
And what if it all goes wrong? JCA will not assess economics or make value judgments on behalf of individual countries, and the assessments are non-binding. However, it will amount to a relative effectiveness assessment versus comparators, which may not go in favor of the treatment being assessed. We know from experience that HTA bodies take note of assessments in other countries. Therefore, the impact of an unfavorable pan-European assessment could be huge. JCA is likely to be a challenge for manufacturers. It will impact some products in 2025, and we need to get it right. It is important to understand the benefits of JCA in order to change.
Considering the challenges, timing is going to be key to success with the JCA process.
Gaming out timing is critical to success
While submitting the JCA dossier during the EMA review process will be a challenge, it gives manufacturers an opportunity to optimize some of their processes. For example, it will encourage companies to consider involvement of market access and HEOR teams much earlier in the drug development process –- the HTA process (or at least part of it) will be conducted alongside the regulatory process in Europe, which means that, when planning for evidence generation, both processes need to be considered.
Since July 2017, EUnetHTA and the EMA have offered parallel consultations for manufacturer companies on evidence generation plans -– this is conducted prior to the pivotal trials for a new product, while phase 2 and 3 trials are still at the planning stage. The objective of these parallel consultations, or joint scientific consultations (JSCs), as they are now known, is to help generate optimal and robust evidence to satisfy both regulators and HTA bodies. It should be noted that there is a finite number of JSC slots available and there are specific eligibility criteria for JSC requests, including:
- unmet medical need for the product
- being first in class
- a large potential impact on patients, public health, or healthcare systems
- a significant cross-border dimension
- and major E.U.-wide added value or clinical research priorities
These criteria can make it difficult to secure a slot.
Even if a company does not wish to participate in a JSC, or if a treatment is not eligible for JSC, there is a compelling argument for considering HTA requirements much earlier in the drug development process. For example, with earlier HTA engagement, clinical trials and real-world studies could potentially be designed to incorporate specific comparators, endpoints, or subgroups relevant to HTA that may not have been necessary from a regulatory perspective but may satisfy regulatory needs. Of course, it may make pre-launch evidence generation planning more complex, and sometimes, it may not be feasible to incorporate all endpoints, comparators, or subgroups that may be relevant to HTA, but at the very least, it facilitates informed planning and decision making.
“We have been working with DRG/Clarivate on a high-profile orphan drug in rare diseases. We have engaged them to lead our International HTA Network to ensure that we submitted high quality HTA Dossiers in Europe. They have demonstrated strong organizational skills, eye for detail and ability to manage external consultancies and deal with internal stakeholders, assertively and diplomatically. “
Early planning related to the communication of treatment value will also become increasingly important with JCA. While economics will come later in the process, companies will need to develop a strategy and value narrative in preparation for JCA submission that will still be applicable during country-specific economic evaluations and pricing negotiations -– this is likely to involve early economic modelling to prepare for the economic scenarios. It will be important to consider relevant messaging for individual countries, but the narrative running through the JCA dossier must be more general global (or at least pan-European).
JCA, RWD and RWE
On a more granular level, predicting PICOs internally in order to plan for evidence generation activities will be important. The key to success with JCA will be early planning and collaboration. Companies really need to be thinking about how their various teams engage at different points in the drug development process and, particularly for market access and HEOR teams, this will need to be much earlier, when during evidence generation planning for pivotal trials.
Real-world evidence is used increasingly frequently in HTA, enabling a more robust critical assessment of technologies and can validate whether the study population and clinical context of a RCT is reflective of clinical practice. RWE offers enormous potential to inform multiple aspects of HTA, including:
- Epidemiology
- Current management
- Clinical and economic outcomes
However there remain challenges for using RWE in HTA, including skepticism about the validity of evidence and that evidence generation timing may not be synchronized with HTA and pricing bodies’ agendas.
Strong partnership among all stakeholders and pragmatic use of existing data alongside clinical evidence provided by companies are key success factors.
Case study: using RWD to inform a cost-effectiveness model to demonstrate the benefits of an inhaled antibiotic for managing a chronic pulmonary infection.
A large pharma company completed a head-to-head trial comparing their inhalation solution using a nebulizer handset with an existing treatment for a chronic pulmonary infection. In the trial, the new inhalation solution demonstrated both clinical and economic benefits compared with the existing standard of care, showing improved lung function and reduced risk of hospitalization.
The client required a model to communicate the value of the inhalation solution and nebulizer handset to national payers in order to demonstrate the cost effectiveness of their new treatment compared to standard of care for inclusion as part of their HTA submissions.
To meet these requirements, a compelling economic model was needed to evaluate the cost-effectiveness of a new inhalation solution administered via a nebulizer compared with standard of care.
Insights generated using Clarivate’s RWD ecosystem of patient-level records enabled the quantification of key areas of differentiation (improved lung function, reduced HCRU) compared with standard of care.
These insights were used inform a cost-effectiveness model to demonstrate the benefit of reduced drug, hospitalization and lung transplantation costs.
This model supported a national HTA submission, and country adaptations were created to aid the client’s local affiliates. This solution:
- Empowered our partner to demonstrate the clinical and economic value of the new inhalation solution and nebulizer system as an alternative to standard of care, in compelling format that was both robust and commercially and strategically optimal
- Provided a user-friendly and flexible cost-effectiveness analysis with scenario and sensitivity analyses to explore potential scenarios and uncertainty around key input data, and support for country adaptations of the model
- Delivered impactful messaging around the value of the product to key stakeholders
Does RWD/RWE influence HTA decisions and market access?
HTA Bodies acknowledge that RWD/RWE should become an essential component of HTA processes; their availability allows questions left open by the initial submission package to be answered.
But generating good evidence is challenging. It challenges the IT infrastructure of each health system, which in each country has its own history contingent on its welfare model, to its supply of healthcare (public or private/public mix), and to its national governance (centralization vs decentralization).
The burden of providing controlled evidence for a new treatment is borne by companies, while for RWD/RWE, it is shared between them and HTA bodies. This leads to an additional administrative burden. The time and skills needed to provide good evidence should be considered.
If you would like to discuss your 2024 plans with any of our R&D, commercialization or technology enablement teams, please connect with our experts here.
This post was written by Ruth Howells and Oliver Blandy.
Ruth Howells is Head of Health Technology Assessment (HTA) at Clarivate. Ruth is responsible for our Global HTA services and has been involved in the development and strategic direction of HTAs, contributing in-depth knowledge of procedural processes. Full bio here.
Oliver Blandy is a Senior Consultant at Clarivate. Oliver provides epidemiology and real-world data insights and solutions to meet the business needs of decision makers within the pharma and biotech industries, working across multiple therapeutic areas including cancer, infectious disease, and rare diseases.


